

Computational Chemistry
Main interests
- Valence bond theory (VB theory) to aid the understanding of the structure and bonding of small, simple non-metal molecules and
- Molecular orbital theory (MO theory) to predict and rationalize the structure and bonding of molecules, as well as to predict the stability of high energy density materials (HEDMs).
There has been a resurgence of interest in the use of valence bond (VB) methods to study the electronic structure of molecules. With regard to the provision of primitive patterns of understanding of electronic structure , VB theory, with localized or two-center bonds in the VB structures for triatomic or larger systems, has many conceptual advantages over qualitative molecular orbital (MO) theory, which uses delocalized bonds via multicenter MOs.
In our research group, we give attention primarily to one type of qualitative VB theory, namely the increased-valence theory, with application to a variety of azid-, nitro- and fluorine-containing molecules. This theory represents a natural extension of the familiar Lewis-type VB theory used by many chemists.
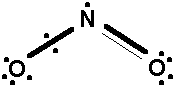
Fig. 1. Increased Lewis structure of the NO2 radical.
An example of the usefulness of VB theory is for the description of molecules such as Ozone, O3 where a long bond structure has been shown through VB calculations to be of considerable importance in the resonance scheme (fig. 2). Calculation of the VB-weights of particular Lewis structures has recently become more accessible by new programmes which are still in development. We are using these new programmes to describe more accurately the importance of resonance in small, simple molecules such as N5+ and ClO2.
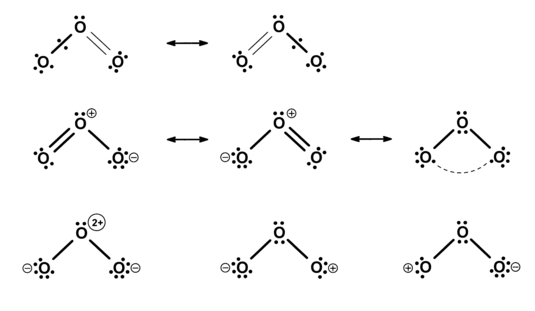
Fig. 2. Resonance structures for ozone in its ground state.
For a review of the current use of VB theory see: R. D. Harcourt, T. M. Klapötke, J. Fluorine Chem. 2003, 123(1), 5 - 20.
We are currently using ab initio calculation not only to rationalize the structure and bonding in compounds, but also for more novel purposes such as to obtain an appreciation for the initial decomposition behavior of energetic materials by calculating possible reaction pathways using either ab initio or density functional theory (DFT). A further interesting application found by Politzer et al. and extensively used by Rice et al. as well as in our research group, is computing the pattern of the electrostatic potential on the surface of a molecule, which can in general, be related to the sensitivity of the bulk material.
Typically, for organic molecules in general, the negative regions cover a smaller portion of the total surface area but are significantly stronger (in terms of average magnitudes) than the positive ones. On the other hand, in the case of energetic molecules, the positive regions are still larger but now also stronger in magnitude than the negative. It was also possible to show that impact sensitivities can be expressed as a function of the extent of this anomalous reversal of the strengths of the positive and negative surface potentials. Fig. 3 shows the calculated electrostatic potentials of tetrazole azide and of phenyltetrazole azide for comparison.
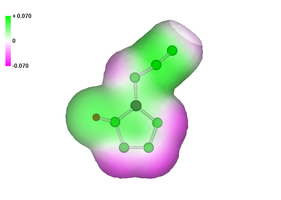
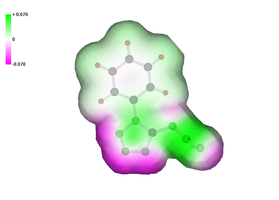
Fig. 3. Calculated electrostatic potentials for the highly explosive tetrazole azide, HCN7 and less explosive phenyltetrazole azide.

Quantum Chemical Methods in Main-Group Chemistry